Research
Agent-based evolutionary modeling enables exploration of the big-picture how and why behind incredible biological capability for adaptation, complexity, and novelty. This field of work, known as “digital evolution,” knits interdisciplinary connections between computer science and evolutionary biology to design and study digital processes and structures that capture lifelike properties. Biologically-inspired techniques leveraging evolution as an algorithm can often produce good solutions to hard real-world problems. They also provide a useful model system to study difficult questions in evolutionary theory.
My work focuses on understanding organisms’ adaptation to the evolutionary process itself (“evolvability”) and on developing methodology to simulate larger-scale digital artificial life systems, particularly with respect to high-performance computing and digital multicellularity. I am particularly passionate about bringing research into practice by building reusable software that advances the field.
You can find more details about research projects I’m involved in below. Selected highlights from my publications are available on my Professional Works page.
 PCA visualization of gene regulation activity in DISHTINY.
PCA visualization of gene regulation activity in DISHTINY.
Studying how artificial evolutionary systems can continually produce novel artifacts of increasing complexity has proven to be a rich vein for practical, scientific, philosophical, and artistic innovations. Unfortunately, existing computational artificial life systems appear constrained by practical limitations on simulation scale. While by no means certain, the idea that orders-of-magnitude increases in compute power will open up qualitatively different possibilities with respect to open-ended evolution is well founded.
Until fundamental changes to computing technology transpire, scaling up artificial life compute power will require taking advantage of parallel and distributed computing systems. Modern high-performance scientific computing clusters appear perhaps the best target to start down this path.
Unlike most existing applications of distributed computing in digital evolution, open-ended evolution researchers must prioritize dynamic interactions among distributed simulation elements. Ecologies, co-evolutionary dynamics, and social behavior all necessitate such dynamic interactions. The question of how to design artificial life simulations and engineer artifical life software at scale will be paramount for the field.
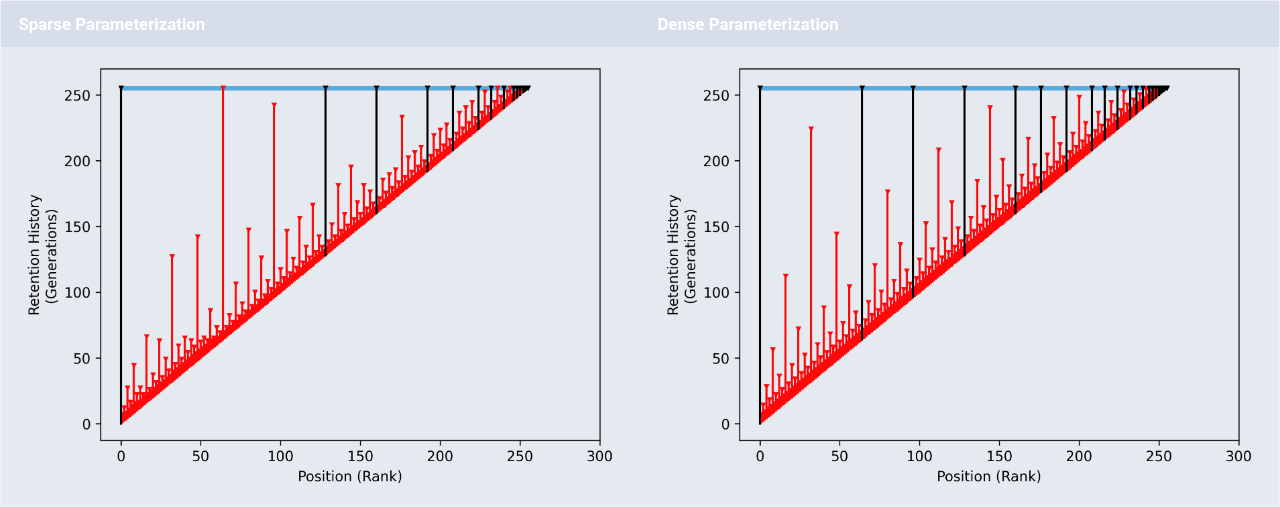 Retention visualization for hereditary stratigraphy policy.
Retention visualization for hereditary stratigraphy policy.
The capability to detect phylogenetic cues within digital evolution has become increasingly necessary in both applied and scientific contexts. These cues unlock post hoc insight into evolutionary history — particularly with respect to ecology and selection pressure — but also can be harnessed to drive digital evolution algorithms as they unfold. However, parallel and distributed evaluation complicates, among other concerns, maintenance of an evolutionary record. Existing phylogenetic record keeping requires inerrant and complete collation of birth and death reports within a centralized data structure. Such perfect tracking approaches are brittle to data loss or corruption and impose communication overhead.
A phylogenetic inference approach, as opposed to phylogenetic tracking, has potential to improve scalability and robustness. Under such a model, history is estimated from comparison of available extant genomes — aligning with the familiar paradigm of phylogenetic work in wet biology. However, this raises the question of how best to design digital genomes to facilitate phylogenetic inference.
This work introduces a new technique, called hereditary stratigraphy, that works by attaching a set of immutable historical “checkpoints” — referred to as strata — as an annotation on evolving genomes.
Checkpoints can be strategically discarded to reduce annotation size at the cost of increasing inference uncertainty.
An accompanying software library, hstrat, provides a plug-and-play implementation of hereditary stratigraphy that can be incorporated into any digital evolution system.
Publications & Software
View at Publisher
| Authors | Matthew Andres Moreno, Connor Yang, Emily Dolson, Luis Zaman |
| Date | April 16th, 2024 |
| DOI | 10.48550/arXiv.2404.10861 |
| Venue | arXiv |
Abstract
Continuing improvements in computing hardware are poised to transform capabilities for in silico modeling of cross-scale phenomena underlying major open questions in evolutionary biology and artificial life, such as transitions in individuality, eco-evolutionary dynamics, and rare evolutionary events. Emerging ML/AI-oriented hardware accelerators, like the 850,000 processor Cerebras Wafer Scale Engine (WSE), hold particular promise. However, practical challenges remain in conducting informative evolution experiments that efficiently utilize these platforms’ large processor counts. Here, we focus on the problem of extracting phylogenetic information from agent-based evolution on the WSE platform. This goal drove significant refinements to decentralized in silico phylogenetic tracking, reported here. These improvements yield order-of-magnitude performance improvements. We also present an asynchronous island-based genetic algorithm (GA) framework for WSE hardware. Emulated and on-hardware GA benchmarks with a simple tracking-enabled agent model clock upwards of 1 million generations a minute for population sizes reaching 16 million agents. We validate phylogenetic reconstructions from these trials and demonstrate their suitability for inference of underlying evolutionary conditions. In particular, we demonstrate extraction, from wafer-scale simulation, of clear phylometric signals that differentiate runs with adaptive dynamics enabled versus disabled. Together, these benchmark and validation trials reflect strong potential for highly scalable agent-based evolution simulation that is both efficient and observable. Developed capabilities will bring entirely new classes of previously intractable research questions within reach, benefiting further explorations within the evolutionary biology and artificial life communities across a variety of emerging high-performance computing platforms.
BibTeX
@misc{moreno2024trackable,
doi={10.48550/arXiv.2404.10861},
url={https://arxiv.org/abs/2404.10861},
title={Trackable Agent-based Evolution Models at Wafer Scale},
author={Matthew Andres Moreno and Connor Yang and Emily Dolson and Luis Zaman},
year={2024},
eprint={2404.10861},
archivePrefix={arXiv},
primaryClass={cs.NE}
}
Citation
Moreno, M. A., Yang, C., Dolson, E., & Zaman, L. (2024). Trackable Agent-based Evolution Models at Wafer Scale. arXiv preprint arXiv:2404.10861.
Supporting Materials
View at Publisher
| Authors | Matthew Andres Moreno, Santiago Rodriguez Papa, Emily Dolson |
| Date | March 3rd, 2024 |
| DOI | 10.48550/arXiv.2403.00246 |
| Venue | arXiv |
Abstract
Since the advent of modern bioinformatics, the challenging, multifaceted problem of reconstructing phylogenetic history from biological sequences has hatched perennial statistical and algorithmic innovation. Studies of the phylogenetic dynamics of digital, agent-based evolutionary models motivate a peculiar converse question: how to best engineer tracking to facilitate fast, accurate, and memory-efficient lineage reconstructions? Here, we formally describe procedures for phylogenetic analysis in both serial and distributed computing scenarios. With respect to the former, we demonstrate reference-counting-based pruning of extinct lineages. For the latter, we introduce a trie-based phylogenetic reconstruction approach for “hereditary stratigraphy” genome annotations. This process allows phylogenetic relationships between genomes to be inferred by comparing their similarities, akin to reconstruction of natural history from biological DNA sequences. Phylogenetic analysis capabilities significantly advance distributed agent-based simulations as a tool for evolutionary research, and also benefit application-oriented evolutionary computing. Such tracing could extend also to other digital artifacts that proliferate through replication, like digital media and computer viruses.
BibTeX
@misc{moreno2024analysis,
doi={10.48550/arXiv.2403.00246},
url={https://arxiv.org/abs/2403.00246},
title={Analysis of Phylogeny Tracking Algorithms for Serial and Multiprocess Applications},
author={Matthew Andres Moreno and Santiago Rodriguez Papa and Emily Dolson},
year={2024},
eprint={2403.00246},
archivePrefix={arXiv},
primaryClass={cs.DS}
}
Citation
Moreno, M. A., Rodriguez Papa, S., & Dolson, E. (2024). Analysis of Phylogeny Tracking Algorithms for Serial and Multiprocess Applications. arXiv preprint arXiv:2403.00246.
Supporting Materials
View at Publisher
| Authors | Matthew Andres Moreno, Santiago Rodriguez Papa, Emily Dolson |
| Date | March 3rd, 2024 |
| DOI | 10.48550/arXiv.2403.00266 |
| Venue | arXiv |
Abstract
Data stream algorithms tackle operations on high-volume sequences of read-once data items. Data stream scenarios include inherently real-time systems like sensor networks and financial markets. They also arise in purely-computational scenarios like ordered traversal of big data or long-running iterative simulations. In this work, we develop methods to maintain running archives of stream data that are temporally representative, a task we call “stream curation.” Our approach contributes to rich existing literature on data stream binning, which we extend by providing stateless (i.e., non-iterative) curation schemes that enable key optimizations to trim archive storage overhead and streamline processing of incoming observations. We also broaden support to cover new trade-offs between curated archive size and temporal coverage. We present a suite of five stream curation algorithms that span O(n), O(logn), and O(1) orders of growth for retained data items. Within each order of growth, algorithms are provided to maintain even coverage across history or bias coverage toward more recent time points. More broadly, memory-efficient stream curation can boost the data stream mining capabilities of low-grade hardware in roles such as sensor nodes and data logging devices.
BibTeX
@misc{moreno2024algorithms,
doi = {10.48550/arXiv.2403.00266},
url = {https://arxiv.org/abs/2403.00246},
title={Algorithms for Efficient, Compact Online Data Stream Curation},
author={Matthew Andres Moreno and Santiago Rodriguez Papa and Emily Dolson},
year={2024},
eprint={2403.00266},
archivePrefix={arXiv},
primaryClass={cs.DS}
}
Citation
Moreno, M. A., Rodriguez Papa, S., & Dolson, E. (2024). Algorithms for Efficient, Compact Online Data Stream Curation. arXiv preprint arXiv:2403.00266.
Supporting Materials
View at Publisher
| Authors | Matthew Andres Moreno |
| Date | February 18th, 2024 |
| Venue | Genetic Programming Theory and Practice XX |
Abstract
The structure of relatedness among members of an evolved population tells much of its evolutionary history. In application-oriented evolutionary computation (EC), such phylogenetic information can guide algorithm selection and tuning. Although traditional direct tracking approaches provide the perfect phylogenetic record, sexual recombination complicates management and analysis of this data. Taking inspiration from biological science, this work explores a reconstruction-based approach that uses end-state genetic information to estimate phylogenetic history after the fact. We apply recently-developed “hereditary stratigraphy” genome annotations to lineages with sexual recombination to design devices germane to species phylogenies and gene trees. As shown through a series of validation experiments, proposed instrumentation can discern genealogical history, population size changes, and selective sweeps. Fully decentralized by nature, these methods afford new observability at scale, in particular, for distributed EC systems. Such capabilities anticipate continued growth of computational resources available to EC. Accompanying open source software aims to expedite application of reconstruction-based phylogenetic analysis where pertinent.
BibTeX
@incollection{moreno2024methods,
author = {Moreno, Matthew Andres},
editor = {Winkler, Stephan
and Trujillo, Leonardo
and Ofria, Charles
and Hu, Ting},
title = {Methods for Rich Phylogenetic Inference Over Distributed Sexual Populations},
booktitle = {Genetic Programming Theory and Practice XX},
year = 2024,
pages = {125--141},
publisher = {Springer International Publishing},
isbn = {978-981-99-8413-8},
doi = {10.1007/978-981-99-8413-8_7},
url = {https://doi.org/10.1007/978-981-99-8413-8_7},
}
Citation
Moreno, M.A. (2024). Methods for Rich Phylogenetic Inference Over Distributed Sexual Populations. In: Winkler, S., Trujillo, L., Ofria, C., Hu, T. (eds) Genetic Programming Theory and Practice XX. Genetic and Evolutionary Computation. Springer, Singapore. https://doi.org/10.1007/978-981-99-8413-8_7
| Authors | Emily Dolson, Santiago Rodriguez-Papa, Matthew Andres Moreno |
| Date | August 10th, 2023 |
| DOI | 10.21105/joss.04866 |
| Venue | Journal of Open Source Software |
Abstract
In silico evolution instantiates the processes of heredity, variation, and differential reproductive success (the three “ingredients” for evolution by natural selection) within digital populations of computational agents. Consequently, these populations undergo evolution, and can be used as virtual model systems for studying evolutionary dynamics. This experimental paradigm — used across biological modeling, artificial life, and evolutionary computation — complements research done using in vitro and in vivo systems by enabling the user to conduct experiments that would be impossible in the lab or field [@dolsonDigitalEvolutionEcology2021]. One key benefit is complete, exact observability. For example, it is possible to perfectly record the full set of parent-child relationships over the history of a population, yielding precise and accurate phylogenies (ancestry trees). This information reveals the sequences of events behind gain, loss, or maintenance of specific traits, and also facilitates making inferences about the underlying evolutionary dynamics of a given system.
The Phylotrack project provides libraries for tracking and analyzing phylogenies in in silico evolution. The project is composed of 1) Phylotracklib: a header-only C++ library, developed under the umbrella of the Empirical project, and 2) Phylotrackpy: a Python wrapper around Phylotracklib, created with Pybind11. Both components supply a public-facing API to attach phylogenetic tracking to digital evolution systems, as well as a stand-alone interface for measuring a variety of popular phylogenetic topology metrics. The underlying algorithm design prioritizes efficiency, allowing Phylotrack to support large agent populations with rapid generational turnover. The underlying C++ implementation ensures fast, memory-efficient performance, with multiple explicit features (e.g., phylogeny pruning and abstraction, etc.) for reducing the memory footprint of phylogenetic information.
BibTeX
in review
Citation
in review
View at Publisher
| Authors | Matthew Andres Moreno, Emily Dolson, Santiago Rodriguez-Papa |
| Date | July 24th, 2023 |
| DOI | 10.1162/isal_a_00694 |
| Venue | The 2023 Conference on Artificial Life |
Abstract
As digital evolution systems grow in scale and complexity, observing and interpreting their evolutionary dynamics will become increasingly challenging. Distributed and parallel computing, in particular, introduce obstacles to maintaining the high level of observability that makes digital evolution a powerful experimental tool. Phylogenetic analyses represent a promising tool for drawing inferences from digital evolution experiments at scale. Recent work has introduced promising techniques for decentralized phylogenetic inference in parallel and distributed digital evolution systems. However, foundational phylogenetic theory necessary to apply these techniques to characterize evolutionary dynamics is lacking. Here, we lay the groundwork for practical applications of distributed phylogenetic tracking in three ways: 1) we present an improved technique for reconstructing phylogenies from tunably-precise genome annotations, 2) we begin the process of identifying how the signatures of various evolutionary dynamics manifest in phylogenetic metrics, and 3) we quantify the impact of reconstruction-induced imprecision on phylogenetic metrics. We find that selection pressure, spatial structure, and ecology have distinct effects on phylogenetic metrics, although these effects are complex and not always intuitive. We also find that, while low-resolution phylogenetic reconstructions can bias some phylogenetic metrics, high-resolution reconstructions recapitulate them faithfully.
BibTeX
@inproceedings{moreno2023toward,
author = {Moreno, Matthew Andres and Dolson, Emily and Rodriguez-Papa, Santiago},
title = "{Toward Phylogenetic Inference of Evolutionary Dynamics at Scale}",
volume = {ALIFE 2023: Ghost in the Machine: Proceedings of the 2023 Artificial Life Conference},
series = {ALIFE 2023: Ghost in the Machine: Proceedings of the 2023 Artificial Life Conference},
pages = {79},
year = {2023},
month = {07},
doi = {10.1162/isal_a_00694},
url = {https://doi.org/10.1162/isal\_a\_00694},
eprint = {https://direct.mit.edu/isal/proceedings-pdf/isal/35/79/2149068/isal\_a\_00694.pdf},
}
Citation
Matthew Andres Moreno, Emily Dolson, Santiago Rodriguez-Papa; July 24–28, 2023. “Toward Phylogenetic Inference of Evolutionary Dynamics at Scale.” Proceedings of the ALIFE 2023: Ghost in the Machine: Proceedings of the 2023 Artificial Life Conference. ALIFE 2023: Ghost in the Machine: Proceedings of the 2023 Artificial Life Conference. Online. (pp. 79). ASME. https://doi.org/10.1162/isal_a_00694
| Authors | Matthew Andres Moreno, Emily Dolson, Charles Ofria |
| Date | November 7th, 2022 |
| DOI | 10.21105/joss.04866 |
| Venue | Journal of Open Source Software |
Abstract
Digital evolution systems instantiate evolutionary processes over populations of virtual agents in silico. These programs can serve as rich experimental model systems. Insights from digital evolution experiments expand evolutionary theory, and can often directly improve heuristic optimization techniques . Perfect observability, in particular, enables in silico experiments that would be otherwise impossible in vitro or in vivo. Notably, availability of the full evolutionary history (phylogeny) of a given population enables very powerful analyses.
As a slow but highly parallelizable process, digital evolution will benefit greatly by continuing to capitalize on profound advances in parallel and distributed computing, particularly emerging unconventional computing architectures. However, scaling up digital evolution presents many challenges. Among these is the existing centralized perfect-tracking phylogenetic data collection model, which is inefficient and difficult to realize in parallel and distributed contexts. Here, we implement an alternative approach to tracking phylogenies across vast and potentially unreliable hardware networks.
BibTeX
@article{moreno2022hstrat,
doi = {10.21105/joss.04866},
url = {https://doi.org/10.21105/joss.04866},
year = {2022},
publisher = {The Open Journal},
volume = {7},
number = {80},
pages = {4866},
author = {Matthew Andres Moreno and Emily Dolson and Charles Ofria},
title = {hstrat: a Python Package for phylogenetic inference on distributed digital evolution populations},
journal = {Journal of Open Source Software}
}
Citation
Moreno et al., (2022). hstrat: a Python Package for phylogenetic inference on distributed digital evolution populations. Journal of Open Source Software, 7(80), 4866, https://doi.org/10.21105/joss.04866
View at Publisher
| Authors | Alexander Lalejini, Matthew Andres Moreno, Charles Ofria |
| Date | July 19th, 2022 |
| DOI | 10.1145/3520304.3534060 |
| Venue | The Genetic and Evolutionary Computation Conference |
Abstract
This Hot-off-the-Press paper summarizes our recently published work, “Tag-based regulation of modules in genetic programming improves context-dependent problem solving,” published in Genetic Programming and Evolvable Machines [1]. We introduce and experimentally demonstrate tag-based genetic regulation, a genetic programming (GP) technique that allows programs to dynamically adjust which code modules to express. Tags are evolvable labels that provide a flexible naming scheme for referencing code modules. Tag-based regulation extends tag-based naming schemes to allow programs to “promote” and “repress” code modules to alter module execution patterns. We find that tag-based regulation improves problem-solving success on problems where programs must adjust how they respond to current inputs based on prior inputs; indeed, some of these problems could not be solved until regulation was added. We also identify scenarios where the correct response to an input does not change over time, rendering tag-based regulation an unnecessary functionality that can sometimes impede evolution. Broadly, tag-based regulation adds to our repertoire of techniques for evolving more dynamic computer programs and can easily be incorporated into existing tag-enabled GP systems.
BibTeX
@inproceedings{lalejini2022tag,
author = {Lalenini, Alexander and Moreno, Matthew Andres and Ofria, Charles},
title = {Tag-based Module Regulation for Genetic Programming},
year = {2022},
isbn = {9781450392686},
publisher = {Association for Computing Machinery},
address = {New York, NY, USA},
url = {https://doi.org/10.1145/3520304.3534060},
doi = {10.1145/3520304.3534060},
booktitle = {Proceedings of the Genetic and Evolutionary Computation Conference Companion},
pages = {25-26},
numpages = {2},
keywords = {gene regulation, genetic programming, SignalGP, automatic program synthesis, tag-based referencing},
location = {Boston, Massachusetts},
series = {GECCO '22}
}
Citation
Alexander Lalejini, Matthew Andres Moreno, and Charles Ofria. 2022. Tag-based Module Regulation for Genetic Programming. In Proceedings of the Genetic and Evolutionary Computation Conference Companion (GECCO ‘22). Association for Computing Machinery, New York, NY, USA, 25–26. https://doi.org/10.1145/3520304.3534060
Supporting Materials
View at Publisher
| Authors | Matthew Andres Moreno, Emily Dolson, Charles Ofria |
| Date | May 13th, 2022 |
| DOI | 10.1145/3520304.3533937 |
| Venue | The Genetic and Evolutionary Computation Conference |
Abstract
Phylogenetic analyses can also enable insight into evolutionary and ecological dynamics such as selection pressure and frequency dependent selection in digital evolution systems. Traditionally digital evolution systems have recorded data for phylogenetic analyses through perfect tracking where each birth event is recorded in a centralized data structures. This approach, however, does not easily scale to distributed computing environments where evolutionary individuals may migrate between a large number of disjoint processing elements. To provide for phylogenetic analyses in these environments, we propose an approach to infer phylogenies via heritable genetic annotations rather than directly track them. We introduce a “hereditary stratigraphy” algorithm that enables efficient, accurate phylogenetic reconstruction with tunable, explicit trade-offs between annotation memory footprint and reconstruction accuracy. This approach can estimate, for example, MRCA generation of two genomes within 10% relative error with 95% confidence up to a depth of a trillion generations with genome annotations smaller than a kilobyte. We also simulate inference over known lineages, recovering up to 85.70% of the information contained in the original tree using a 64-bit annotation.
BibTeX
@inproceedings{moreno2022hereditary_gecco,
author = {Moreno, Matthew Andres and Dolson, Emily and Ofria, Charles},
title = {Hereditary Stratigraphy: Genome Annotations to Enable Phylogenetic Inference over Distributed Populations},
year = {2022},
isbn = {9781450392686},
publisher = {Association for Computing Machinery},
address = {New York, NY, USA},
url = {https://doi.org/10.1145/3520304.3533937},
doi = {10.1145/3520304.3533937},
booktitle = {Proceedings of the Genetic and Evolutionary Computation Conference Companion},
pages = {65–66},
numpages = {2},
keywords = {phylogenetics, decentralized algorithms, genetic algorithms, digital evolution, genetic programming},
location = {Boston, Massachusetts},
series = {GECCO '22}
}
Citation
Matthew Andres Moreno, Emily Dolson, and Charles Ofria. 2022. Hereditary stratigraphy: genome annotations to enable phylogenetic inference over distributed populations. In Proceedings of the Genetic and Evolutionary Computation Conference Companion (GECCO ‘22). Association for Computing Machinery, New York, NY, USA, 65–66. https://doi.org/10.1145/3520304.3533937
View at Publisher
| Authors | Matthew Andres Moreno, Emily Dolson, Charles Ofria |
| Date | May 13th, 2022 |
| DOI | 10.1162/isal_a_00550 |
| Venue | The 2022 Conference on Artificial Life |
Abstract
Phylogenies provide direct accounts of the evolutionary trajectories behind evolved artifacts in genetic algorithm and artificial life systems. Phylogenetic analyses can also enable insight into evolutionary and ecological dynamics such as selection pressure and frequency-dependent selection. Traditionally, digital evolution systems have recorded data for phylogenetic analyses through perfect tracking where each birth event is recorded in a centralized data structure. This approach, however, does not easily scale to distributed computing environments where evolutionary individuals may migrate between a large number of disjoint processing elements. To provide for phylogenetic analyses in these environments, we propose an approach to enable phylogenies to be inferred via heritable genetic annotations rather than directly tracked. We introduce a “hereditary stratigraphy” algorithm that enables efficient, accurate phylogenetic reconstruction with tunable, explicit trade-offs between annotation memory footprint and reconstruction accuracy. In particular, we demonstrate an approach that enables estimation of the most recent common ancestor (MRCA) between two individuals with fixed relative accuracy irrespective of lineage depth while only requiring logarithmic annotation space complexity with respect to lineage depth This approach can estimate, for example, MRCA generation of two genomes within 10% relative error with 95% confidence up to a depth of a trillion generations with genome annotations smaller than a kilobyte. We also simulate inference over known lineages, recovering up to 85.70% of the information contained in the original tree using 64-bit annotations.
BibTeX
@inproceedings{moreno2022hereditary,
author = {Moreno, Matthew Andres and Dolson, Emily and Ofria, Charles},
title = "{Hereditary Stratigraphy: Genome Annotations to Enable Phylogenetic Inference over Distributed Populations}",
volume = {ALIFE 2022: The 2022 Conference on Artificial Life},
series = {ALIFE 2022: The 2022 Conference on Artificial Life},
year = {2022},
month = {07},
doi = {10.1162/isal_a_00550},
url = {https://doi.org/10.1162/isal\_a\_00550},
note = {64},
eprint = {https://direct.mit.edu/isal/proceedings-pdf/isal/34/64/2035363/isal\_a\_00550.pdf},
}
Citation
Matthew Andres Moreno, Emily Dolson, Charles Ofria; July 18–22, 2022. “Hereditary Stratigraphy: Genome Annotations to Enable Phylogenetic Inference over Distributed Populations.” Proceedings of the ALIFE 2022: The 2022 Conference on Artificial Life. ALIFE 2022: The 2022 Conference on Artificial Life. Online. (pp. 64). ASME. https://doi.org/10.1162/isal_a_00550
View at Publisher
| Authors | Matthew Andres Moreno, Emily Dolson, Charles Ofria |
| Date | January 1st, 2022 |
| Venue | Python package published via PyPI |
hstrat enables phylogenetic inference on distributed digital evolution populations.
BibTeX
@article{moreno2022hereditary,
author = {Moreno, Matthew Andres and Dolson, Emily and Ofria, Charles},
doi = {https://doi.org/10.1162/isal_a_00550},
journal = {Proceedings of the ALIFE 2022: The 2022 Conference on Artificial Life},
month = {7},
pages = {64--74},
title = {{Hereditary Stratigraphy: Genome Annotations to Enable Phylogenetic Inference over Distributed Populations}},
volume = {Proceedings of the ALIFE 2022: The 2022 Conference on Artificial Life},
year = {2022}
}
Citation
Moreno, M. A., Dolson, E., & Ofria, C. (2022). Hereditary Stratigraphy: Genome Annotations to Enable Phylogenetic Inference over Distributed Populations. Proceedings of the ALIFE 2022: The 2022 Conference on Artificial Life, Proceedings of the ALIFE 2022: The 2022 Conference on Artificial Life(), 64–74. https://doi.org/https://doi.org/10.1162/isal_a_00550
 Sequence of multicellular phenotypes observed in a DISHTINY experiment.
Sequence of multicellular phenotypes observed in a DISHTINY experiment.
Evolutionary transitions occur when previously-independent replicating entities unite to form more complex individuals. The necessary conditions and evolutionary mechanisms for these transitions to arise continue to be fruitful targets of scientific interest. Likewise, the relationship of such transitions to continuing generation of novelty, complexity, and adaptation remains an open question.
This work uses a digital model of multicellularity to study a range of fraternal transitions in populations of open-ended self-replicating computer programs. These digital cells are allowed to form and replicate kin groups by selectively adjoining or expelling daughter cells. This model provides an opportunity study group-level traits that are characteristic of a fraternal transition. These include reproductive division of labor, resource sharing within kin groups, resource investment in offspring groups, asymmetrical behaviors mediated by messaging, morphological patterning, and adaptive apoptosis.
Ongoing work with this model seeks to tease apart the interplay between novelty, complexity, and adaptation in evolution, with early results suggesting a loose, sometimes divergent, relationship.
Publications & Software
View at Publisher
| Authors | Matthew Andres Moreno |
| Date | April 16th, 2024 |
| DOI | 10.48550/arXiv.2404.10854 |
| Venue | arXiv |
Abstract
Complexity is a signature quality of interest in artificial life systems. Alongside other dimensions of assessment, it is common to quantify genome sites that contribute to fitness as a complexity measure. However, limitations to the sensitivity of fitness assays in models with implicit replication criteria involving rich biotic interactions introduce the possibility of difficult-to-detect “cryptic” adaptive sites, which contribute small fitness effects below the threshold of individual detectability or involve epistatic redundancies. Here, we propose three knockout-based assay procedures designed to quantify cryptic adaptive sites within digital genomes. We report initial tests of these methods on a simple genome model with explicitly configured site fitness effects. In these limited tests, estimation results reflect ground truth cryptic sequence complexities well. Presented work provides initial steps toward development of new methods and software tools that improve the resolution, rigor, and tractability of complexity analyses across alife systems, particularly those requiring expensive in situ assessments of organism fitness.
BibTeX
@misc{moreno2024methods,
doi={10.48550/arXiv.2404.10854},
url={https://arxiv.org/abs/2404.10854},
title={Methods to Estimate Cryptic Sequence Complexity},
author={Matthew Andres Moreno},
year={2024},
eprint={2404.10854},
archivePrefix={arXiv},
primaryClass={q-bio.PE}
}
Citation
Moreno, M. A. (2024). Methods to Estimate Cryptic Sequence Complexity. arXiv preprint arXiv:2404.10854.
Supporting Materials
View at Publisher
| Authors | Matthew Andres Moreno, Charles Ofria |
| Date | May 13th, 2022 |
| DOI | 10.3389/fevo.2022.750837 |
| Venue | Frontiers in Ecology and Evolution |
Abstract
Evolutionary transitions occur when previously-independent replicating entities unite to form more complex individuals. Such transitions have profoundly shaped natural evolutionary history and occur in two forms: fraternal transitions involve lower-level entities that are kin (e.g., transitions to multicellularity or to eusocial colonies), while egalitarian transitions involve unrelated individuals (e.g., the origins of mitochondria). The necessary conditions and evolutionary mechanisms for these transitions to arise continue to be fruitful targets of scientific interest. Here, we examine a range of fraternal transitions in populations of open-ended self-replicating computer programs. These digital cells were allowed to form and replicate kin groups by selectively adjoining or expelling daughter cells. The capability to recognize kin-group membership enabled preferential communication and cooperation between cells. We repeatedly observed group-level traits that are characteristic of a fraternal transition. These included reproductive division of labor, resource sharing within kin groups, resource investment in offspring groups, asymmetrical behaviors mediated by messaging, morphological patterning, and adaptive apoptosis. We report eight case studies from replicates where transitions occurred and explore the diverse range of adaptive evolved multicellular strategies.
BibTeX
@article{moreno2022exploring,
author={Moreno, Matthew Andres and Ofria, Charles},
title={Exploring Evolved Multicellular Life Histories in a Open-Ended Digital Evolution System},
journal={Frontiers in Ecology and Evolution},
volume={10},
year={2022},
url={https://www.frontiersin.org/articles/10.3389/fevo.2022.750837},
doi={10.3389/fevo.2022.750837},
issn={2296-701X}
}
Citation
Moreno MA and Ofria C (2022) Exploring Evolved Multicellular Life Histories in a Open-Ended Digital Evolution System. Front. Ecol. Evol. 10:750837. doi: 10.3389/fevo.2022.750837
View at Publisher
| Authors | Matthew Andres Moreno, Santiago Rodriguez Papa, Charles Ofria |
| Date | July 22nd, 2021 |
| Venue | The Fourth Workshop on Open-Ended Evolution (OEE4) |
Abstract
Continuing generation of novelty, complexity, and adaptation are well-established as core aspects of open-ended evolution. However, the manner in which these phenomena relate remains an area of great theoretical interest. It is yet to be firmly established to what extent these phenomena are coupled and by what means they interact. In this work, we track the co-evolution of novelty, complexity, and adaptation in a case study from a simulation system designed to study the evolution of digital multicellularity. In this case study, we describe ten qualitatively distinct multicellular morphologies, several of which exhibit asymmetrical growth and distinct life stages. We contextualize the evolutionary history of these morphologies with measurements of complexity and adaptation. Our case study suggests a loose, sometimes divergent, relationship can exist among novelty, complexity, and adaptation.
BibTeX
@inproceedings{moreno2021case,
author = {Moreno, Matthew Andres and Papa, Santiago Rodriguez and Ofria, Charles},
title = {Case Study of Novelty, Complexity, and Adaptation in a Multicellular System},
year = {2021},
url = {http://workshops.alife.org/oee4/papers/moreno-oee4-camera-ready.pdf},
booktitle = {OEE4: The Fourth Workshop on Open-Ended Evolution},
numpages = {9},
location = {Prague, Czech Republic}
}
Citation
Matthew Andres Moreno, Santiago Rodriguez Papa and Charles Ofria. 2021. Case Study of Novelty, Complexity, and Adaptation in a Multicellular System. OEE4: The Fourth Workshop on Open-Ended Evolution.
| Authors | Matthew Andres Moreno, Santiago Rodriguez Papa, Katherine Perry, Charles Ofria |
| Date | January 1st, 2020 |
| Venue | header-only C++ library |
C++ library for digital evolution simulations studying digital multicellularity and fraternal major evolutionary transitions in individuality.
View at Publisher
| Authors | Matthew Andres Moreno, Charles Ofria |
| Date | May 1st, 2019 |
| DOI | 10.1162/artl_a_00284 |
| Venue | Artificial Life |
Abstract
The emergence of new replicating entities from the union of simpler entities characterizes some of the most profound events in natural evolutionary history. Such transitions in individuality are essential to the evolution of the most complex forms of life. Thus, understanding these transitions is critical to building artificial systems capable of open-ended evolution. Alas, these transitions are challenging to induce or detect, even with computational organisms. Here, we introduce the DISHTINY (Distributed Hierarchical Transitions in Individuality) platform, which provides simple cell-like organisms with the ability and incentive to unite into new individuals in a manner that can continue to scale to subsequent transitions. The system is designed to encourage these transitions so that they can be studied: organisms that coordinate spatiotemporally can maximize the rate of resource harvest, which is closely linked to their reproductive ability. We demonstrate the hierarchical emergence of multiple levels of individuality among simple cell-like organisms that evolve parameters for manually designed strategies. During evolution, we observe reproductive division of labor and close cooperation among cells, including resource-sharing, aggregation of resource endowments for propagules, and emergence of an apoptosis response to somatic mutation. Many replicate populations evolved to direct their resources toward low-level groups (behaving like multicellular individuals), and many others evolved to direct their resources toward high-level groups (acting as larger-scale multicellular individuals).
BibTeX
@article{moreno2019toward,
author = {Moreno, Matthew Andres and Ofria, Charles},
title = "{Toward Open-Ended Fraternal Transitions in Individuality}",
journal = {Artificial Life},
volume = {25},
number = {2},
pages = {117-133},
year = {2019},
month = {05},
issn = {1064-5462},
doi = {10.1162/artl_a_00284},
url = {https://doi.org/10.1162/artl\_a\_00284},
eprint = {https://direct.mit.edu/artl/article-pdf/25/2/117/1896700/artl\_a\_00284.pdf},
}
Citation
Matthew Andres Moreno, Charles Ofria; Toward Open-Ended Fraternal Transitions in Individuality. Artif Life 2019; 25 (2): 117–133. doi: https://doi.org/10.1162/artl_a_00284
View at Publisher
| Authors | Matthew Andres Moreno, Charles Ofria |
| Date | July 22nd, 2018 |
| Venue | The Third Workshop on Open-Ended Evolution (OEE3) |
Abstract
The emergence of new replicating entities from the union of existing entities represent some of the most profound events in natural evolutionary history. Facilitating such evolutionary transitions in individuality is essential to the derivation of the most complex forms of life. As such, understanding these transitions is critical for building artificial systems capable of open-ended evolution. Alas, these transitions are challenging to induce or detect, even with computational organisms. Here, we introduce the DISHTINY (DIStributed Hierarchical Transitions in IndividualitY) platform, which provides simple cell-like organisms with the ability and incentive to unite into new individuals in a manner that can continue to scale to subsequent transitions. The system is designed to encourage these transitions so that they can be studied: organisms that coordinate spatiotemporally can maximize the rate of resource harvest, which is closely linked to their reproductive ability. We demonstrate the hierarchical emergence of multiple levels of individuality among simple cell-like organisms that evolve parameters for manually-designed strategies. During evolution, we observe reproductive division of labor and close cooperation between cells, including resource-sharing, aggregation of resource endowments for propagules, and emergence of an apoptosis response to somatic mutation. While a few replicate populations evolved selfish behaviors, many evolved to direct their resources toward low-level groups (behaving like multi-cellular individuals), and many others evolved to direct their resources toward high-level groups (acting as larger-scale multi-cellular individuals). Finally, we demonstrated that genotypes that encode higher-level individuality consistently outcompete those that encode lower-level individuality.
BibTeX
@inproceedings{moreno2018understanding,
author = {Moreno, Matthew Andres and Ofria, Charles},
title = {Understanding Fraternal Transitions in Individuality},
year = {2018},
url = {http://workshops.alife.org/oee3/papers/moreno-oee3-final.pdf},
booktitle = {OEE3: The Third Workshop on Open-Ended Evolution},
numpages = {8},
location = {Tokyo, Japan}
}
Citation
Matthew Andres Moreno and Charles Ofria. 2018. Understanding Fraternal Transitions in Individuality. OEE3: The Third Workshop on Open-Ended Evolution.
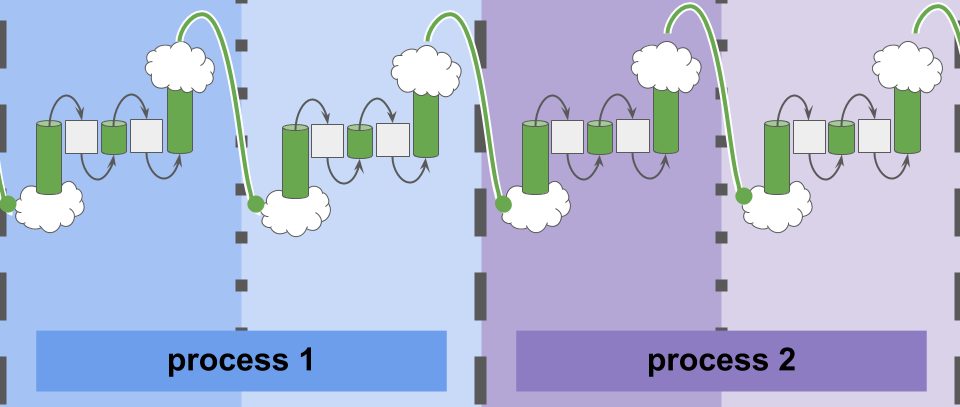 Cartoon illustration of communication between simulation elements in experiment with Conduit software.
Cartoon illustration of communication between simulation elements in experiment with Conduit software.
The parallel and distributed processing capacity of high-performance computing (HPC) clusters continues to grow rapidly and enable profound scientific and industrial innovations. These advances in hardware capacity and economy afford great opportunity, but also pose a serious challenge: developing approaches to effectively harness it.
Software and hardware that relaxes guarantees of correctness and determinism — a so-called ``best-effort model’’ — have been shown to improve speed. This work distills best-effort communication from the larger issue of best-effort computing. Specifically, we investigate the implications of relaxing synchronization and message delivery requirements. Such a best-effort approach meets the challenges of heterogenous, varying (i.e., due to power management), and generally lower communication bandwidth (relative to compute) expected on future HPC hardware. Notably, such a model presents the possibility of runtime adaptation to effectively utilize available resources given the particular ratio of compute and communication capability at any one moment in any one rack.
Complex biological organisms exhibit characteristic best-effort properties: trillions of cells interact asynchronously while overcoming all but the most extreme failures in a noisy world. As such, bio-inspired algorithms present strong potential to benefit from best-effort communication strategies.
Much exciting work on best-effort computing has incorporated bespoke experimental hardware. However, existing software libraries for traditional HPC hardware do not typically explicitly expose a convenient best-effort communication interface for such work. This work introduces the Conduit library, which facilitates best-effort communication between parallel and distributed processes on existing, commercially-available hardware.
Publications & Software
View at Publisher
| Authors | Matthew Andres Moreno, Connor Yang, Emily Dolson, Luis Zaman |
| Date | April 16th, 2024 |
| DOI | 10.48550/arXiv.2404.10861 |
| Venue | arXiv |
Abstract
Continuing improvements in computing hardware are poised to transform capabilities for in silico modeling of cross-scale phenomena underlying major open questions in evolutionary biology and artificial life, such as transitions in individuality, eco-evolutionary dynamics, and rare evolutionary events. Emerging ML/AI-oriented hardware accelerators, like the 850,000 processor Cerebras Wafer Scale Engine (WSE), hold particular promise. However, practical challenges remain in conducting informative evolution experiments that efficiently utilize these platforms’ large processor counts. Here, we focus on the problem of extracting phylogenetic information from agent-based evolution on the WSE platform. This goal drove significant refinements to decentralized in silico phylogenetic tracking, reported here. These improvements yield order-of-magnitude performance improvements. We also present an asynchronous island-based genetic algorithm (GA) framework for WSE hardware. Emulated and on-hardware GA benchmarks with a simple tracking-enabled agent model clock upwards of 1 million generations a minute for population sizes reaching 16 million agents. We validate phylogenetic reconstructions from these trials and demonstrate their suitability for inference of underlying evolutionary conditions. In particular, we demonstrate extraction, from wafer-scale simulation, of clear phylometric signals that differentiate runs with adaptive dynamics enabled versus disabled. Together, these benchmark and validation trials reflect strong potential for highly scalable agent-based evolution simulation that is both efficient and observable. Developed capabilities will bring entirely new classes of previously intractable research questions within reach, benefiting further explorations within the evolutionary biology and artificial life communities across a variety of emerging high-performance computing platforms.
BibTeX
@misc{moreno2024trackable,
doi={10.48550/arXiv.2404.10861},
url={https://arxiv.org/abs/2404.10861},
title={Trackable Agent-based Evolution Models at Wafer Scale},
author={Matthew Andres Moreno and Connor Yang and Emily Dolson and Luis Zaman},
year={2024},
eprint={2404.10861},
archivePrefix={arXiv},
primaryClass={cs.NE}
}
Citation
Moreno, M. A., Yang, C., Dolson, E., & Zaman, L. (2024). Trackable Agent-based Evolution Models at Wafer Scale. arXiv preprint arXiv:2404.10861.
Supporting Materials
View at Publisher
| Authors | Matthew Andres Moreno, Charles Ofria |
| Date | November 23rd, 2022 |
| DOI | 10.48550/arXiv.2211.10897 |
| Venue | arXiv |
Abstract
Here, we test the performance and scalability of fully-asynchronous, best-effort communication on existing, commercially-available HPC hardware.
A first set of experiments tested whether best-effort communication strategies can benefit performance compared to the traditional perfect communication model. At high CPU counts, best-effort communication improved both the number of computational steps executed per unit time and the solution quality achieved within a fixed-duration run window.
Under the best-effort model, characterizing the distribution of quality of service across processing components and over time is critical to understanding the actual computation being performed. Additionally, a complete picture of scalability under the best-effort model requires analysis of how such quality of service fares at scale. To answer these questions, we designed and measured a suite of quality of service metrics: simulation update period, message latency, message delivery failure rate, and message delivery coagulation. Under a lower communication-intensivity benchmark parameterization, we found that median values for all quality of service metrics were stable when scaling from 64 to 256 process. Under maximal communication intensivity, we found only minor – and, in most cases, nil – degradation in median quality of service.
In an additional set of experiments, we tested the effect of an apparently faulty compute node on performance and quality of service. Despite extreme quality of service degradation among that node and its clique, median performance and quality of service remained stable.
BibTeX
@misc{moreno2022best,
doi = {10.48550/ARXIV.2211.10897},
url = {https://arxiv.org/abs/2211.10897},
author = {Moreno, Matthew Andres and Ofria, Charles},
keywords = {Distributed, Parallel, and Cluster Computing (cs.DC), FOS: Computer and information sciences, FOS: Computer and information sciences},
title = {Best-Effort Communication Improves Performance and Scales Robustly on Conventional Hardware},
publisher = {arXiv},
year = {2022},
copyright = {arXiv.org perpetual, non-exclusive license}
}
Citation
Moreno, M. A., & Ofria, C. (2022). Best-Effort Communication Improves Performance and Scales Robustly on Conventional Hardware. arXiv preprint arXiv:2211.10897.
Supporting Materials
View at Publisher
| Authors | Matthew Andres Moreno, Santiago Rodriguez Papa, Charles Ofria |
| Date | May 21st, 2021 |
| DOI | 10.1145/3449726.3463205 |
| Venue | ACM Workshop on Parallel and Distributed Evolutionary Inspired Methods |
Abstract
Developing software to effectively take advantage of growth in parallel and distributed processing capacity poses significant challenges. Traditional programming techniques allow a user to assume that execution, message passing, and memory are always kept synchronized. However, maintaining this consistency becomes increasingly costly at scale. One proposed strategy is “best-effort computing”, which relaxes synchronization and hardware reliability requirements, accepting nondeterminism in exchange for efficiency. Although many programming languages and frameworks aim to facilitate software development for high performance applications, existing tools do not directly provide a prepackaged best-effort interface. The Conduit C++ Library aims to provide such an interface for convenient implementation of software that uses best-effort inter-thread and inter-process communication. Here, we describe the motivation, objectives, design, and implementation of the library. Benchmarks on a communication-intensive graph coloring problem and a compute-intensive digital evolution simulation show that Conduit’s best-effort model can improve scaling efficiency and solution quality, particularly in a distributed, multi-node context.
BibTeX
@inproceedings{moreno2021conduit,
author = {Moreno, Matthew Andres and Papa, Santiago Rodriguez and Ofria, Charles},
title = {Conduit: A C++ Library for Best-Effort High Performance Computing},
year = {2021},
isbn = {9781450383516},
publisher = {Association for Computing Machinery},
address = {New York, NY, USA},
url = {https://doi.org/10.1145/3449726.3463205},
doi = {10.1145/3449726.3463205},
booktitle = {Proceedings of the Genetic and Evolutionary Computation Conference Companion},
pages = {1795–1800},
numpages = {6},
keywords = {high performance computing, best-effort computing},
location = {Lille, France},
series = {GECCO '21}
}
Citation
Matthew Andres Moreno, Santiago Rodriguez Papa, and Charles Ofria. 2021. Conduit: a C++ library for best-effort high performance computing. In Proceedings of the Genetic and Evolutionary Computation Conference Companion (GECCO ‘21). Association for Computing Machinery, New York, NY, USA, 1795–1800. https://doi.org/10.1145/3449726.3463205
View at Publisher
| Authors | Matthew Andres Moreno, Santiago Rodriguez Papa, Charles Ofria |
| Date | March 12th, 2021 |
| Venue | The 6th International Workshop on Modeling and Simulation of and by Parallel and Distributed Systems (MSPDS 2020) |
Abstract
Developing software to effectively take advantage of growth in parallel and distributed processing capacity poses significant challenges. Best-effort computing models, which relax synchronization requirements, have been proposed as a strategy to overcome challenges harness high performance computing at extreme scale. Although many programming languages and frameworks aim to facilitate software development for high performance applications, existing prevalent tools do not expose an explicit best-effort interface. The Conduit C++ Library aims to provide a convenient interface for best-effort inter-thread and inter-process communication. Here, we describe the motivation, objectives, design, and implementation of the library.
BibTeX
@inproceedings{moreno2021conduit_hpcs,
author = {Moreno, Matthew Andres and Papa, Santiago Rodriguez and Ofria, Charles},
title = {Conduit: A C++ Library for Best-Effort High Performance Computing},
year = {2021},
booktitle = {The 6th International Workshop on Modeling and Simulation of and by Parallel and Distributed Systems (MSPDS 2020)},
numpages = {2},
keywords = {high performance computing, best-effort computing},
location = {Barcelona, Sapin},
series = {HPCS 2021}
}
Citation
Matthew Andres Moreno, Santiago Rodriguez Papa and Charles Ofria. 2021. Conduit: A C++ Library for Best-Effort High Performance Computing. MSPDS 2020: The 6th International Workshop on Modeling and Simulation of and by Parallel and Distributed Systems.
Supporting Materials
| Authors | Matthew Andres Moreno, Santiago Rodriguez Papa, Charles Ofria |
| Date | January 1st, 2020 |
| Venue | header-only C++ library |
C++ library that wraps intra-thread, inter-thread, and inter-process communication in a uniform, modular, object-oriented interface, with a focus on asynchronous high-performance computing applications.
 Group photo of participants in 2020 and 2021 Workshops for Avida-ED Software Development.
Group photo of participants in 2020 and 2021 Workshops for Avida-ED Software Development.
Open source software supercharges the rate of scientific progress and the applied praxis of those advances. Interdisciplinary fields like artificial life, which thrive due to contributions from those without formal training in computing such as biologists and mathematicians, especially benefit from published applications and software packages.
Open source devleopment of research software holds core priority within my work, with particular emphasis on maximizing its broader usefullness to the broader community outside of its original context. Teaching and mentorship also constitues a core aspect of this work, empowering researchers with development capabilities and promoting best practices in the community. I led the 2020 and 2021 Workshop for Avida-ED Software Development, which paired 27 early-career participants mentored 10 week hands-on projects, most related to writing, testing, and documenting software. I have also mentored five undergraduates on scientific software development projects.
 Include graph for DISHTINY software.
Include graph for DISHTINY software.
Packaging and distribution of software multiplies the impact of research, both by opening the door to follow-on research within the scientific community and by facilitating direct real-world applications. However, realizing this goal requires special attention to organization, documentation, and reliability. Many of my research projects are organized so to maximize contribution of general-purpose library software back to the community. This usually involves adding software features to an existing project or publishing a standalone Python or C++ library.
Publications & Software
View at Publisher
| Authors | Matthew Andres Moreno |
| Date | December 22nd, 2023 |
| Venue | Python package published via PyPI |
add zoom indicators, insets, and magnified panels to matplotlib/seaborn visualizations with ease!
BibTeX
@software{moreno2023outset,
author = {Matthew Andres Moreno},
title = {mmore500/outset},
month = dec,
year = 2023,
publisher = {Zenodo},
doi = {10.5281/zenodo.10426106},
url = {https://doi.org/10.5281/zenodo.10426106}
}
Citation
Matthew Andres Moreno. (2023). mmore500/outset. Zenodo. https://doi.org/10.5281/zenodo.10426106
Supporting Materials
View at Publisher
| Authors | Matthew Andres Moreno, Emily Dolson |
| Date | January 1st, 2022 |
| Venue | Python package published via PyPI |
phylotrackpy is a Python phylogeny tracker.
View at Publisher
| Authors | Matthew Andres Moreno |
| Date | January 1st, 2022 |
| Venue | Python package published via PyPI |
opytional makes working with values that might be None safer and easier.
View at Publisher
| Authors | Matthew Andres Moreno |
| Date | January 1st, 2022 |
| Venue | Python package published via PyPI |
interval-search provides predicate-based binary and doubling search implementations.
View at Publisher
| Authors | Matthew Andres Moreno, Emily Dolson, Charles Ofria |
| Date | January 1st, 2022 |
| Venue | Python package published via PyPI |
hstrat enables phylogenetic inference on distributed digital evolution populations.
BibTeX
@article{moreno2022hereditary,
author = {Moreno, Matthew Andres and Dolson, Emily and Ofria, Charles},
doi = {https://doi.org/10.1162/isal_a_00550},
journal = {Proceedings of the ALIFE 2022: The 2022 Conference on Artificial Life},
month = {7},
pages = {64--74},
title = {{Hereditary Stratigraphy: Genome Annotations to Enable Phylogenetic Inference over Distributed Populations}},
volume = {Proceedings of the ALIFE 2022: The 2022 Conference on Artificial Life},
year = {2022}
}
Citation
Moreno, M. A., Dolson, E., & Ofria, C. (2022). Hereditary Stratigraphy: Genome Annotations to Enable Phylogenetic Inference over Distributed Populations. Proceedings of the ALIFE 2022: The 2022 Conference on Artificial Life, Proceedings of the ALIFE 2022: The 2022 Conference on Artificial Life(), 64–74. https://doi.org/https://doi.org/10.1162/isal_a_00550
View at Publisher
| Authors | Matthew Andres Moreno, Emily Dolson |
| Date | January 1st, 2022 |
| Venue | Python package published via PyPI |
alifedata-phyloinformatics-convert helps apply traditional phyloinformatics software to alife standardized data.
| Authors | Matthew Andres Moreno, Santiago Rodriguez Papa |
| Date | May 26th, 2020 |
| Venue | Workshop for Avida-ED Software Development |
Hands-on, asynchronous 4 day tutorial series covering foundational web development competencies, C++ development with the Empirical library, and compiling for the web with Emscripten.
View at Publisher
| Authors | Matthew Andres Moreno |
| Date | January 1st, 2020 |
| Venue | Python package published via PyPI |
teeplot wrangles your data visualizations out of notebooks for you.
BibTeX
@software{moreno2023teeplot,
author = {Matthew Andres Moreno},
title = {mmore500/teeplot},
month = dec,
year = 2023,
publisher = {Zenodo},
doi = {10.5281/zenodo.10440670},
url = {https://doi.org/10.5281/zenodo.10440670}
}
Citation
Matthew Andres Moreno. (2023). mmore500/teeplot. Zenodo. https://doi.org/10.5281/zenodo.10440670
| Authors | Matthew Andres Moreno, Santiago Rodriguez Papa, Alexander Lalejini, Charles Ofria |
| Date | January 1st, 2020 |
| Venue | header-only C++ library |
A genetic programming implementation designed for large-scale artificial life applications. Organized as a header-only C++ library. Inspired by Alex Lalejini’s SignalGP.
| Authors | Matthew Andres Moreno, Santiago Rodriguez Papa, Charles Ofria |
| Date | January 1st, 2020 |
| Venue | header-only C++ library |
C++ library that wraps intra-thread, inter-thread, and inter-process communication in a uniform, modular, object-oriented interface, with a focus on asynchronous high-performance computing applications.
| Authors | Matthew Andres Moreno, Santiago Rodriguez Papa, Katherine Perry, Charles Ofria |
| Date | January 1st, 2020 |
| Venue | header-only C++ library |
C++ library for digital evolution simulations studying digital multicellularity and fraternal major evolutionary transitions in individuality.
View at Publisher
| Authors | Matthew Andres Moreno |
| Date | January 1st, 2019 |
| Venue | Python package published via PyPI |
keyname helps easily pack and unpack metadata in a filename.
| Date | January 1st, 2018 |
| Venue | header-only C++ library |
Empirical is a library of tools for developing useful, efficient, reliable, and available scientific software. The provided code is header-only and encapsulated into the emp namespace, so it is simple to incorporate into existing projects.
BibTeX
@software{Ofria_Empirical_C_library_2020,
author = {Ofria, Charles and Moreno, Matthew Andres and Dolson, Emily and Lalejini, Alex and Rodriguez Papa, Santiago and Fenton, Jake and Perry, Katherine and Jorgensen, Steven and hoffmanriley and grenewode and Baldwin Edwards, Oliver and Stredwick, Jason and cgnitash and theycallmeHeem and Vostinar, Anya and Moreno, Ryan and Schossau, Jory and Zaman, Luis and djrain},
doi = {10.5281/zenodo.4141943},
license = {MIT},
month = {10},
title = {{Empirical: C++ library for efficient, reliable, and accessible scientific software}},
url = {https://github.com/devosoft/Empirical},
version = {0.0.4},
year = {2020}
}
Citation
Ofria, C., Moreno, M. A., Dolson, E., Lalejini, A., Rodriguez Papa, S., Fenton, J., Perry, K., Jorgensen, S., , H., , G., Baldwin Edwards, O., Stredwick, J., , C., , T., Vostinar, A., Moreno, R., Schossau, J., Zaman, L., & , D. (2020). Empirical: C++ library for efficient, reliable, and accessible scientific software (Version 0.0.4) [Computer software]. https://doi.org/10.5281/zenodo.4141943
Supporting Materials
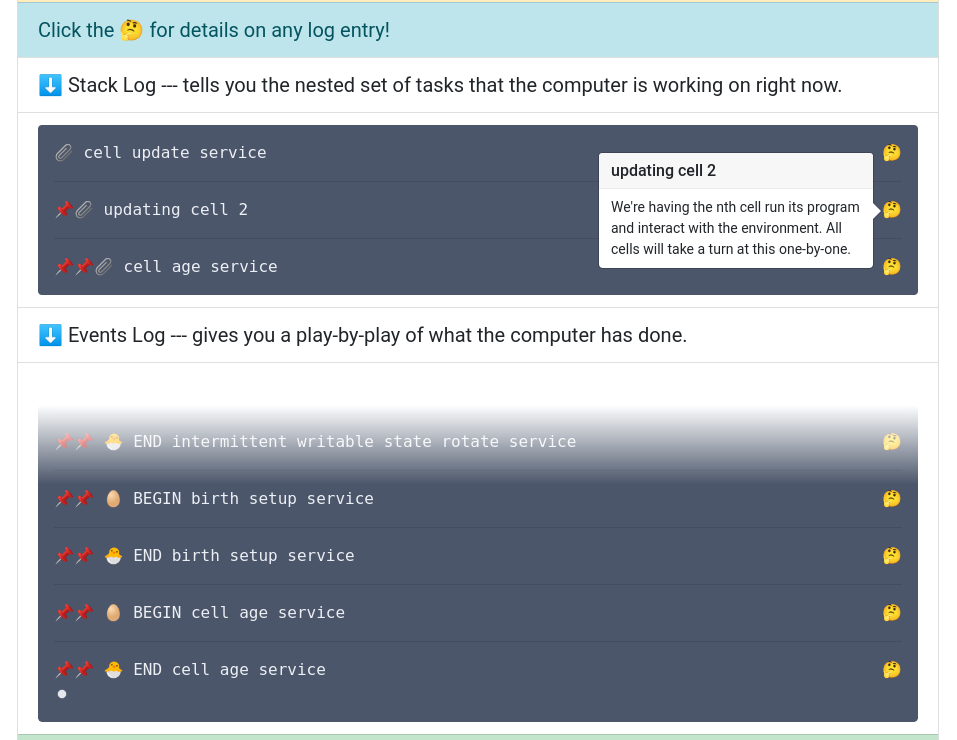 Live execution log for DISHTINY web app.
Live execution log for DISHTINY web app.
Although nominally open sourced, much scientific software is inaccessible in practice because it is prohibitively difficult to use — particularly for members of the general public. This software usually requires users to manually download and install the software, manage complicated software dependencies, and have familiarity with command-line interfaces. Browser-based software with an interactive GUI removes those barriers for both the public and for other scientists.
The Empirical project unlocks the benefit potential of in-browser scientific software by providing a C++ interface to implement HTML GUIs that wrap the pre-existing research version of software, making it easier for researchers to keep the most recent version of scientific software available widely.
Where possibles, I package my experiments & software with Empirical and publish them as interactive in-browser apps like DISHTINY. I also serve as a core contributor on the Empirical project, leading development of re-usable “prefabricated” GUI components and maintaining support for the Emscripten Web Worker API in the core library, among other responsibilities.
Publications & Software
| Authors | Matthew Andres Moreno, Santiago Rodriguez Papa, Katherine Perry, Charles Ofria |
| Date | January 1st, 2020 |
| Venue | header-only C++ library |
C++ library for digital evolution simulations studying digital multicellularity and fraternal major evolutionary transitions in individuality.
| Date | January 1st, 2018 |
| Venue | header-only C++ library |
Empirical is a library of tools for developing useful, efficient, reliable, and available scientific software. The provided code is header-only and encapsulated into the emp namespace, so it is simple to incorporate into existing projects.
BibTeX
@software{Ofria_Empirical_C_library_2020,
author = {Ofria, Charles and Moreno, Matthew Andres and Dolson, Emily and Lalejini, Alex and Rodriguez Papa, Santiago and Fenton, Jake and Perry, Katherine and Jorgensen, Steven and hoffmanriley and grenewode and Baldwin Edwards, Oliver and Stredwick, Jason and cgnitash and theycallmeHeem and Vostinar, Anya and Moreno, Ryan and Schossau, Jory and Zaman, Luis and djrain},
doi = {10.5281/zenodo.4141943},
license = {MIT},
month = {10},
title = {{Empirical: C++ library for efficient, reliable, and accessible scientific software}},
url = {https://github.com/devosoft/Empirical},
version = {0.0.4},
year = {2020}
}
Citation
Ofria, C., Moreno, M. A., Dolson, E., Lalejini, A., Rodriguez Papa, S., Fenton, J., Perry, K., Jorgensen, S., , H., , G., Baldwin Edwards, O., Stredwick, J., , C., , T., Vostinar, A., Moreno, R., Schossau, J., Zaman, L., & , D. (2020). Empirical: C++ library for efficient, reliable, and accessible scientific software (Version 0.0.4) [Computer software]. https://doi.org/10.5281/zenodo.4141943
Supporting Materials
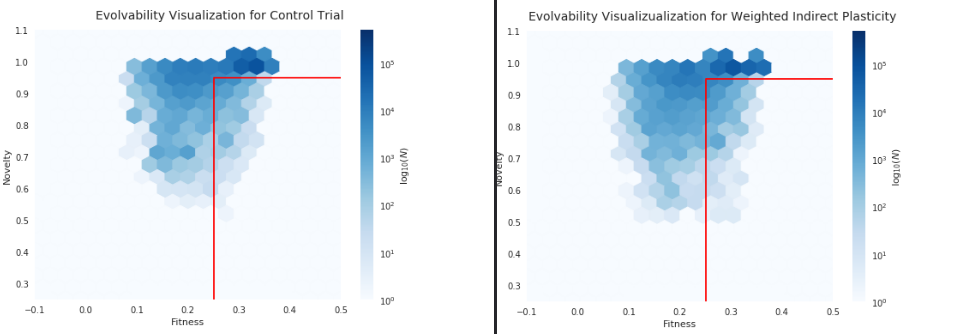 Evolvability signatures from developmental genotype-phenotype map models.
Evolvability signatures from developmental genotype-phenotype map models.
Successful evolutionary search depends on the production of meaningful phenotypic variation that can be inherited by offspring. Without useful heritable variation evolution stagnates. The concept of evolvability describes a population’s capacity to generate useful heritable phenotypic variation is of evolvability. Different evolving systems can exhibit different degrees of evolvability.
Natural systems, in particular, are usually considered to have a capability to continuously generate interesting variation compared to computational systems. Understanding — and replicating – the evolvability of natural evolution is an open problem in computational evolution research.
Evolvability is desirable in artificial evolution systems for practical ends – more evolvable systems will help evolutionary algorithms to tackle sophisticated problems more effectively and efficiently. Understanding evolvability is of great scientific interest for both evolutionary biologists and evolutionary computing researchers, not only for optimization but also with respect to questions related to the evolution of complexity and open-ended evolution.
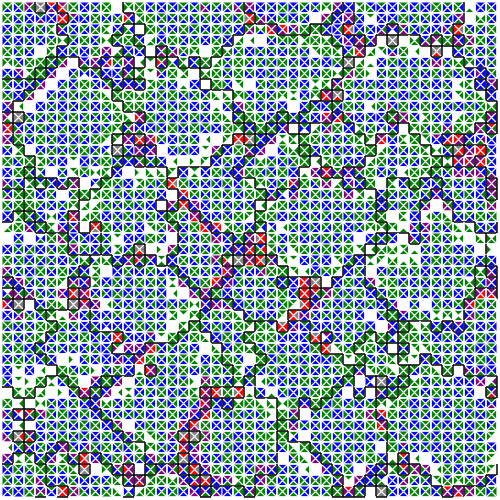 Module expression snapshot in a DISHTINY case study.
Module expression snapshot in a DISHTINY case study.
Genetic programming and artificial life systems commonly use tag matching to decide interactions between system components. However, the implications of criteria used to determine affinity between tags with respect evolutionary dynamics have not been directly studied. Mechanisms to allow reconfiguration of tag interactions at runtime through dynamic regulation remain unexplored, as well.
This line of work explores how that tag-matching processes can influence the rate of adaptive evolution and the quality of evolved solutions. Better understanding of these processes will facilitate more effective incorporation of tag matching into genetic programming and artificial life systems. By showing that tag-matching processes influence connectivity patterns and evolutionary dynamics, our findings also raise fundamental questions about the properties of tag-matching systems in nature.
Publications & Software
View at Publisher
| Authors | Matthew Andres Moreno, Alexander Lalejini, Charles Ofria |
| Date | July 17th, 2023 |
| DOI | 10.1145/3583133.3595834 |
| Venue | The Genetic and Evolutionary Computation Conference |
Abstract
This Hot-off-the-Press paper summarizes our recently published work, “Matchmaker, Matchmaker, Make Me a Match: Geometric, Variational, and Evolutionary Implications of Criteria for Tag Affinity.” This work appeared in Genetic Programming and Evolvable Machines. Genetic programming systems commonly use tag matching to decide interactions between system components. However, the implications of criteria used to determine affinity between tags with respect evolutionary dynamics have not been directly studied. We investigate differences between tag-matching criteria with respect to geometric constraint and variation generated under mutation. In experiments, we find that tag-matching criteria can influence the rate of adaptive evolution and the quality of evolved solutions. Better understanding of the geometric, variational, and evolutionary properties of tag-matching criteria will facilitate more effective incorporation of tag matching into genetic programming systems. By showing that tag-matching criteria influence connectivity patterns and evolutionary dynamics, our findings also raise fundamental questions about the properties of tag-matching systems in nature.
BibTeX
@inproceedings{moreno2023tag,
author = {Moreno, Matthew Andres and Lalejini, Alexander and Ofria, Charles},
title = {Tag Affinity Criteria Influence Adaptive Evolution},
isbn = {9798400701207},
year = {2023},
publisher= {Association for Computing Machinery},
address = {New York, NY, USA},
url = {https://doi.org/10.1145/3583133.3595834},
doi = {10.1145/3583133.3595834},
booktitle= {Proceedings of the Genetic and Evolutionary Computation Conference Companion},
pages = {35-36},
numpages = {2},
keywords = {artificial gene regulatory networks, tag-based referencing, genetic programming, module-based genetic programming, event-driven genetic programming},
location = {Lisbon, Portugal},
series = {GECCO '23}
}
Citation
Matthew Andres Moreno, Alexander Lalejini, and Charles Ofria. 2023. Tag Affinity Criteria Influence Adaptive Evolution. In Proceedings of the Companion Conference on Genetic and Evolutionary Computation (GECCO ‘23 Companion). Association for Computing Machinery, New York, NY, USA, 35–36. https://doi.org/10.1145/3583133.3595834
Supporting Materials
View at Publisher
| Authors | Matthew Andres Moreno, Alexander Lalejini, Charles Ofria |
| Date | March 24th, 2023 |
| DOI | 10.1007/s10710-023-09448-0 |
| Venue | Genetic Programming and Evolvable Machines |
Abstract
Genetic programming and artificial life systems commonly use tag matching to decide interactions between system components. However, the implications of criteria used to determine affinity between tags with respect evolutionary dynamics have not been directly studied. We investigate differences between tag-matching criteria with respect to geometric constraint and variation generated under mutation. In experiments, we find that tag-matching criteria can influence the rate of adaptive evolution and the quality of evolved solutions. Better understanding of the geometric, variational, and evolutionary properties of tag-matching criteria will facilitate more effective incorporation of tag matching into genetic programming and artificial life systems. By showing that tag-matching criteria influence connectivity patterns and evolutionary dynamics, our findings also raise fundamental questions about the properties of tag-matching systems in nature.
BibTeX
@article{moreno2023matchmaker,
author = {Moreno, Matthew Andres and Lalejini, Alexander and Ofria, Charles},
title = {Matchmaker, matchmaker, make me a match: geometric, variational, and evolutionary implications of criteria for tag affinity},
journal = {Genetic Programming and Evolvable Machines},
year = {2023},
month = {Mar},
day = {24},
volume = {24},
number = {1},
pages = {4},
issn = {1573-7632},
doi = {10.1007/s10710-023-09448-0},
url = {https://doi.org/10.1007/s10710-023-09448-0}
}
Citation
Moreno, M.A., Lalejini, A. & Ofria, C. Matchmaker, matchmaker, make me a match: geometric, variational, and evolutionary implications of criteria for tag affinity. Genet Program Evolvable Mach 24, 4 (2023). https://doi.org/10.1007/s10710-023-09448-0
Supporting Materials
View at Publisher
| Authors | Alexander Lalejini, Matthew Andres Moreno, Charles Ofria |
| Date | July 7th, 2021 |
| DOI | 10.1007/s10710-021-09406-8 |
| Venue | Genetic Programming and Evolvable Machines |
Abstract
We introduce and experimentally demonstrate the utility of tag-based genetic regulation, a new genetic programming (GP) technique that allows programs to dynamically adjust which code modules to express. Tags are evolvable labels that provide a flexible mechanism for referencing code modules. Tag-based genetic regulation extends existing tag-based naming schemes to allow programs to “promote” and “repress” code modules in order to alter expression patterns. This extension allows evolution to structure a program as a gene regulatory network where modules are regulated based on instruction executions. We demonstrate the functionality of tag-based regulation on a range of program synthesis problems. We find that tag-based regulation improves problem-solving performance on context-dependent problems; that is, problems where programs must adjust how they respond to current inputs based on prior inputs. Indeed, the system could not evolve solutions to some context-dependent problems until regulation was added. Our implementation of tag-based genetic regulation is not universally beneficial, however. We identify scenarios where the correct response to a particular input never changes, rendering tag-based regulation an unneeded functionality that can sometimes impede adaptive evolution. Tag-based genetic regulation broadens our repertoire of techniques for evolving more dynamic genetic programs and can easily be incorporated into existing tag-enabled GP systems.
BibTeX
@article{lalejini2021tag,
title = {Tag-based regulation of modules in genetic programming improves context-dependent problem solving},
copyright = {All rights reserved},
issn = {1389-2576, 1573-7632},
url = {https://link.springer.com/10.1007/s10710-021-09406-8},
doi = {10.1007/s10710-021-09406-8},
language = {en},
urldate = {2021-07-10},
journal = {Genetic Programming and Evolvable Machines},
volume = {22},
number = {3},
pages = {325--355},
author = {Lalejini, Alexander and Moreno, Matthew Andres and Ofria, Charles},
month = jul,
year = {2021},
}
Citation
Lalejini, A., Moreno, M.A. & Ofria, C. Tag-based regulation of modules in genetic programming improves context-dependent problem solving. Genet Program Evolvable Mach 22, 325–355 (2021). https://doi.org/10.1007/s10710-021-09406-8
Supporting Materials
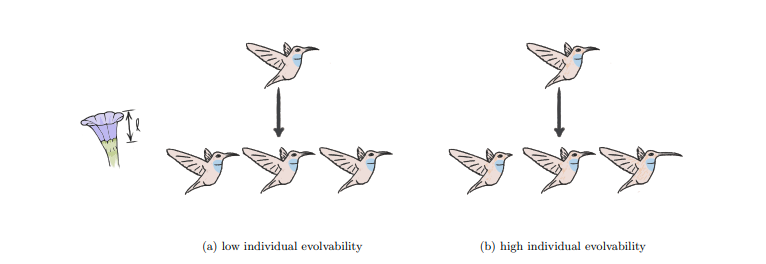 Illustration of selection for evolvability via modularly-varying fitness function.
Illustration of selection for evolvability via modularly-varying fitness function.
Biological organisms exhibit spectacular adaptation to their environments. However, another marvel of biology lurks behind the adaptive traits that organisms exhibit over the course of their lifespans: it is hypothesized that biological organisms also exhibit adaptation to the evolutionary process itself. Although a great deal of fruitful work has explored this idea of “evolvability,” disparate and orthogonal definitions and explanations have splintered in the literature.
This work pursues a review of evolvability theory and a unifying organization of theory, illustrated with examples and experiments from biology and evolutionary computing. It is hoped that a more nuanced and comprehensive understanding of this aspect of evolution will translate to more powerful digital evolution techniques.
Publications & Software
View at Publisher
| Authors | Matthew Andres Moreno |
| Date | December 17th, 2022 |
| Venue | Doctoral Dissertation |
Abstract
Evolutionary transitions occur when previously-independent replicating entities unite to form more complex individuals. Such major transitions in individuality have profoundly shaped complexity, novelty, and adaptation over the course of natural history. Regard for their causes and consequences drives many fundamental questions in biology. Likewise, evolutionary transitions have been highlighted as a hallmark of true open-ended evolution in artificial life. As such, experiments with digital multicellularity promise to help realize computational systems with properties that more closely resemble those of biological systems, ultimately providing insights about the origins of complex life in the natural world and contributing to bio-inspired distributed algorithm design.
Major challenges exist, however, in applying high-performance computing to the dynamic, large-scale digital artificial life simulations required for such work. This dissertation presents two new tools that facilitate such simulations at scale: the Conduit library for best-effort communication and the hstrat (“hereditary stratigraphy”) library, which debuts novel decentralized algorithms to estimate phylogenetic distance between evolving agents.
Most current high-performance computing work emphasizes logical determinism: extra effort is expended to guarantee reliable communication between processing elements. When necessary, computation halts in order to await expected messages. Determinism does enable hardware-independent results and perfect reproducibility, however adopting a best-effort communication model can substantially reduce synchronization overhead and allow dynamic (albeit, potentially lossy) scaling of communication load to fully utilize available resources. We present a set of experiments that test the best-effort communication model implemented by the Conduit library on commercially available high-performance computing hardware. We find that best-effort communication enables significantly better computational performance under high thread and process counts and can achieve significantly better solution quality within a fixed time constraint.
In a similar vein, phylogenetic analysis in digital evolution work has traditionally used a perfect tracking model where each birth event is recorded in a centralized data structure. This approach, however, is difficult scale robustly and efficiently to distributed computing environments where agents may migrate between a dynamic set of disjoint processing elements. To provide for phylogenetic analyses in these environments, we propose an approach to infer phylogenies via heritable genetic annotations. We introduce hereditary stratigraphy, an algorithm that enables tunable trade-offs between annotation memory footprint and accuracy of phylogenetic inference. Simulating inference over known lineages, we recover up to 85% of the information contained in the true phylogeny using only a 64-bit annotation.
We harness these tools in DISHTINY, a distributed digital evolution system designed to study digital organisms as they undergo major evolutionary transitions in individuality. This system allows digital cells to form and replicate kin groups by selectively adjoining or expelling daughter cells. The capability to recognize kin-group membership enables preferential communication and cooperation between cells. We report group-level traits characteristic of fraternal transitions, including reproductive division of labor, resource sharing within kin groups, resource investment in offspring groups, asymmetrical behaviors mediated by messaging, morphological patterning, and adaptive apoptosis. In one detailed case study, we track the co-evolution of novelty, complexity, and adaptation over the evolutionary history of an experiment. We characterize ten qualitatively distinct multicellular morphologies, several of which exhibit asymmetrical growth and distinct life stages. Our case study suggests a loose relationship can exist among novelty, complexity, and adaptation.
The constructive potential inherent in major evolutionary transitions holds great promise for progress toward replicating the capability and robustness of natural organisms. Coupled with shrewd software engineering and innovative model design informed by evolutionary theory, contemporary hardware systems could plausibly already suffice to realize paradigm-shifting advances in open-ended evolution and, ultimately, scientific understanding of major transitions themselves. This work establishes important new tools and methodologies to support continuing progress in this direction.
BibTeX
@phdthesis{moreno2022engineering,
author={Moreno,Matthew A.},
year={2022},
title={Engineering Scalable Digital Models to Study Major Transitions in Evolution},
journal={ProQuest Dissertations and Theses},
pages={379},
note={Copyright - Database copyright ProQuest LLC; ProQuest does not claim copyright in the individual underlying works; Last updated - 2022-12-27},
keywords={Artificial life; Digital evolution; Experimental evolution; High-performance computing; Major transitions in evolution; Simulation; Computer science; Evolution & development; 0984:Computer science; 0412:Evolution and Development},
isbn={9798358499232},
language={English},
url={http://ezproxy.msu.edu/login?url=https://www.proquest.com/dissertations-theses/engineering-scalable-digital-models-study-major/docview/2754890561/se-2},
}
Citation
Moreno, Matthew Andres. 2022. “Engineering Scalable Digital Models to Study Major Transitions in Evolution.” Order No. 29999702, Michigan State University. http://ezproxy.msu.edu/login?url=https://www.proquest.com/dissertations-theses/engineering-scalable-digital-models-study-major/docview/2754890561/se-2.
Supporting Materials
View at Publisher
| Authors | Matthew Andres Moreno |
| Date | April 17th, 2017 |
| Venue | Otis C. Chapman Honors Program Thesis |
Abstract
Biological organisms exhibit spectacular adaptation to their environments. However, another marvel of biology lurks behind the adaptive traits that organisms exhibit over the course of their lifespans: it is hypothesized that biological organisms also exhibit adaptation to the evolutionary process itself. That is, biological organisms are thought to possess traits that facilitate evolution. The term evolvability was coined to describe this type of adaptation. The question of evolvability has special practical relevance to computer science researchers engaged in longstanding efforts to harness evolution as an algorithm for automated design. It is hoped that a more nuanced understanding of biological evolution will translate to more powerful digital evolution techniques. This thesis will present a theoretical overview of evolvability, illustrated with examples from biology and evolutionary computing, and discuss computational experiments probing the relationship between environmental influence on the phenotype and evolvability.
BibTeX
@thesis{moreno2017evolvability,
author={Moreno, Matthew Andres},
title={Evolvability: What Is It and How Do We Get It?},
school={University of Puget Sound},
type={Bachelor's Thesis},
url={http://soundideas.pugetsound.edu/honors_program_theses/22/},
year={2017}
}
Citation
Moreno, Matthew Andres, “Evolvability: What Is It and How Do We Get It?” (2017). Honors Program Theses. 22. https://soundideas.pugetsound.edu/honors_program_theses/22
Supporting Materials
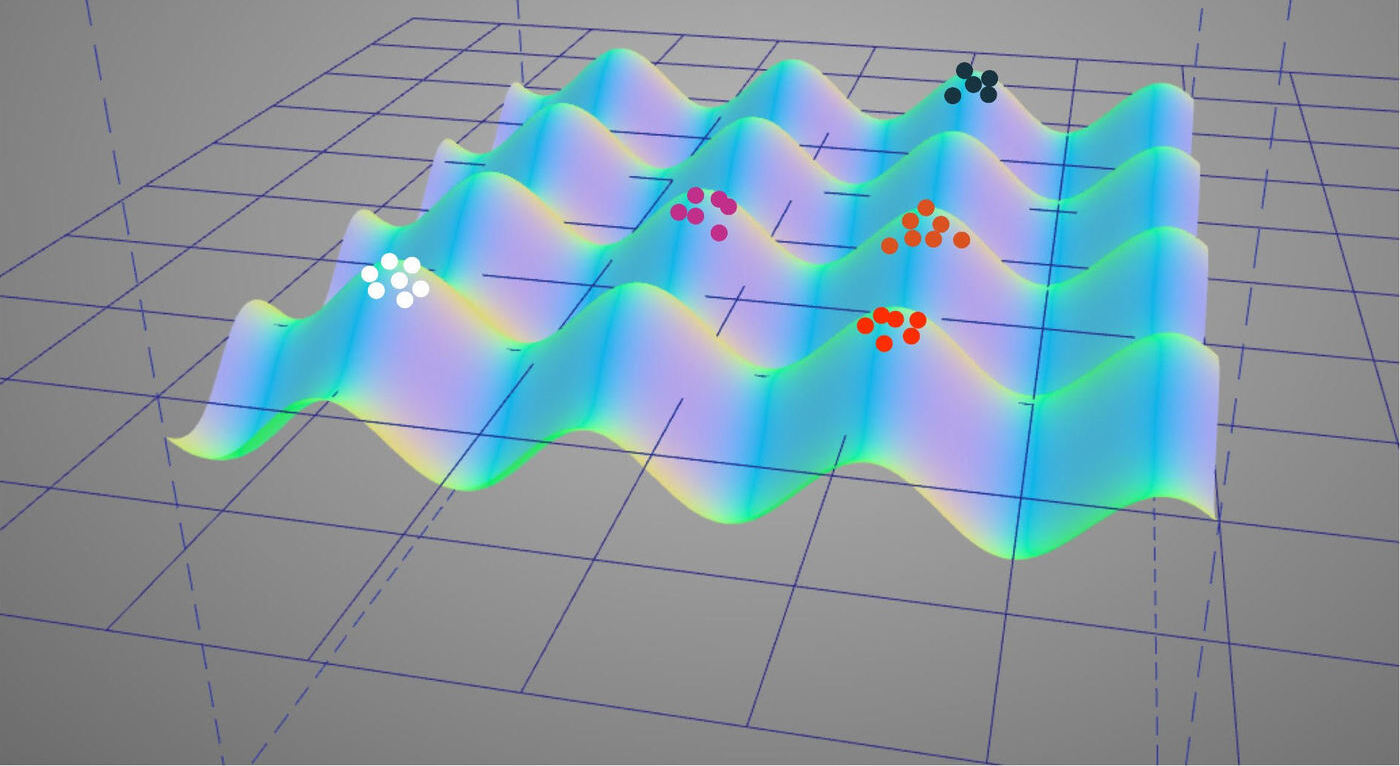 Visualization of rugged fitness landscape suited to denoising autoencoder genotype-phenotype map.
Visualization of rugged fitness landscape suited to denoising autoencoder genotype-phenotype map.
In biology, phenotype refers to an organism’s observable characteristics (morphological, behavioral, physiological, chemical, etc.). Importantly, the phenotype governs an organism’s ability to survive and reproduce — its fitness. Likewise, genotype refers to the heritable information that shapes an organism’s phenotype. This is typically equated with an organism’s DNA content.
The genotype-phenotype map describes the relationship between an organism’s genotype and its phenotype. In biology, this concept is tightly entwined with the process of development, the dynamics through which an organism’s genotype and environment interact to determine its phenotype. As might be expected, the genotype-phenotype map profoundly influences the character of phenotypic variation that is produced under genetic mutation.
Evolutionary search cannot succeed without the production of heritable, viable phenotypic variation, a trait referred to as evolvability. How to engineer genotype-phenotype maps to promote evolvability in digital evolution systems remains a crucial open question.
This work introduces the use of bottlenecked and denoising autoencoders to automatically generate evolvable genotype-phenotype mappings. Autoencoders are algorithms that learn to regurgitate a particular type of input, usually implemented using deep learning. They allow for enhancement of evolvability due to their ability to repair poor-quality solutions and compactly represent the space of high-quality solutions.
Publications & Software
View at Publisher
| Authors | Matthew Andres Moreno, Wolfgang Banzhaf, Charles Ofria |
| Date | July 15th, 2018 |
| DOI | 10.1145/3205455.3205597 |
| Venue | The Genetic and Evolutionary Computation Conference |
Abstract
We present AutoMap, a pair of methods for automatic generation of evolvable genotype-phenotype mappings. Both use an artificial neural network autoencoder trained on phenotypes harvested from fitness peaks as the basis for a genotype-phenotype mapping. In the first, the decoder segment of a bottlenecked autoencoder serves as the genotype-phenotype mapping. In the second, a denoising autoencoder serves as the genotype-phenotype mapping. Automatic generation of evolvable genotype-phenotype mappings are demonstrated on the n-legged table problem, a toy problem that defines a simple rugged fitness landscape, and the Scrabble string problem, a more complicated problem that serves as a rough model for linear genetic programming. For both problems, the automatically generated genotype-phenotype mappings are found to enhance evolvability.
BibTeX
@inproceedings{moreno2018learning,
author = {Moreno, Matthew Andres and Banzhaf, Wolfgang and Ofria, Charles},
title = {Learning an Evolvable Genotype-Phenotype Mapping},
year = {2018},
isbn = {9781450356183},
publisher = {Association for Computing Machinery},
address = {New York, NY, USA},
url = {https://doi.org/10.1145/3205455.3205597},
doi = {10.1145/3205455.3205597},
booktitle = {Proceedings of the Genetic and Evolutionary Computation Conference},
pages = {983–990},
numpages = {8},
keywords = {deep learning, indirect encodings, evolvability, genetic algorithms, adaptive representations, genotype-phenotype map},
location = {Kyoto, Japan},
series = {GECCO '18}
}
Citation
Matthew Andres Moreno, Wolfgang Banzhaf, and Charles Ofria. 2018. Learning an evolvable genotype-phenotype mapping. In Proceedings of the Genetic and Evolutionary Computation Conference (GECCO ‘18). Association for Computing Machinery, New York, NY, USA, 983–990. https://doi.org/10.1145/3205455.3205597